
Antibody Drug Conjugates (ADCs): A guide to Regulatory Clinical Development
Antibody–Drug Conjugates (ADCs): Translating Regulatory Complexity Into Efficient Clinical Development Sandra L. McGuigan MD, MBA
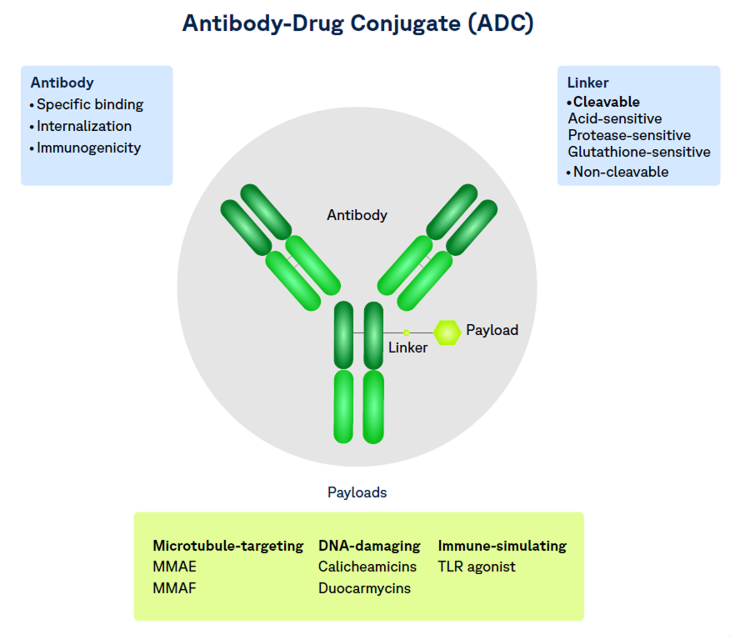
Antibody–drug conjugates (ADCs) are reshaping oncology drug development, offering targeted delivery with chemotherapy‑level potency, which can help reduce off-target side effects. At the same time, ADCs introduce unique clinical and regulatory challenges that can derail timelines if not anticipated early.
In Fortrea’s experience, ADC programs that integrate regulatory expectations early into the development process are positioned to progress more efficiently. This article outlines how sponsors can proactively manage ADC‑specific risks, align protocol design with regulatory expectations, and navigate differences across FDA, EMA, and PMDA.
Why ADCs Require a Different Development Mindset
Unlike traditional biologics or small molecules, ADCs are hybrid products:
- A monoclonal antibody (biologic)
- A highly potent cytotoxic payload (small molecules)
- A linker that governs stability, release, and systemic exposure
Regulators therefore evaluate ADCs through multiple lenses simultaneously—quality, clinical pharmacology, and patient safety. In practice, this means that clinical development decisions are scrutinized not only for efficacy, but for how well they manage ADC‑specific risk.
A Practical ADC Development Checklist--From IND to Approval
IND / CTA: Setting the Foundation for Safe First‑in‑Human Studies
Clinical Development Priorities
- Conservative starting dose selection (often PK- or MABEL-informed)
- Sentinel dosing and staggered escalation
- Extended DLT windows to capture delayed toxicity
- Early and proactive monitoring for ADC class effects (e.g., myelosuppression, ILD/pneumonitis, hepatic toxicity)
Regulatory Expectations For Protocol Design
- Clear rationale linking nonclinical data to starting dose
- Explicit stopping rules tied to payload-related risks
- Early safety biomarkers and imaging where appropriate
- Alignment between dose escalation design and future expansion strategy
Fortrea perspective: Early ADC protocols should already anticipate what regulators will ask at end‑of‑Phase 1, not just what is needed to start dosing.
Phase 1 / Early Phase 2: Dose Optimization Is No Longer Optional
Clinical Pharmacology Is Now Central
Regulators expect ADC programs to evaluate multiple analytes, not just total drug:
- Conjugated ADC
- Total antibody
- Unconjugated (free) payload
Clinical Development Implications
- Expansion cohorts should test more than one clinically relevant dose
- PK/PD sampling must be sufficient to support exposure–response modeling
- Dose modifications should be prespecified and mechanistically justified
- Drug–drug interaction and QTc risk assessments may be required depending on payload
Fortrea perspective: ADC dose finding is not about identifying the maximum tolerated dose—it is about defending the optimal dose for long-term benefit–risk, a core focus of modern regulatory review.
Phase 3: Proving Reproducible Benefit–Risk
Clinical Strategy Considerations
- Clear justification of line of therapy and comparator choice
- Robust safety characterization over time, including cumulative toxicity
- Protocol-defined management of ADC-specific adverse events
- Subgroup analyses informed by target expression or prior therapy
Regulatory Readiness
- Manufacturing process consistency and comparability planning
- Early discussions on accelerated approval vs full approval pathways
- Alignment on post-approval safety commitments
Fortrea perspective: Late-phase ADC development depends as much on operational execution and safety consistency as on headline efficacy.
Real World Evidence Perspective on Antibody–Drug Conjugates
Antibody–drug conjugates (ADCs) are reshaping oncology by combining targeted antibodies with highly potent cytotoxic payloads, but their clinical complexity makes real world evidence (RWE) essential for understanding their true benefit–risk profile. While pivotal trials establish efficacy under controlled conditions, they often underrepresent the older, comorbid, and heavily pre treated patients who receive ADCs in routine practice. As a result, RWE has become critical for assessing generalizability, informing patient selection, and contextualizing trial outcomes.
Real world studies have demonstrated that ADC effectiveness and tolerability are driven as much by molecular design—payload class, linker chemistry, and Fc mediated uptake—as by tumor target. Large post marketing and real-world datasets consistently show distinct toxicity signatures across ADC classes, including ocular toxicity, myelosuppression, and interstitial lung disease (ILD). RWE has clarified earlier onset, higher real world incidence, and substantial mortality associated with ADC related ILD, directly influencing monitoring practices, dose management, and labeling.
RWE can show the difference between trial dosing and real world use, with frequent dose delays, reductions, and discontinuations that shape effectiveness, persistence, and cost outcomes. Comparative real world analyses suggests that outcomes vary more by ADC agent and payload than by indication alone, underscoring the importance of post approval comparative effectiveness research. From a regulatory and lifecycle perspective, ADCs exemplify the growing potential for RWE to fulfill post marketing commitments, support label evolution, and guide clinical adoption. As the ADC pipeline expands toward more complex constructs and earlier line use, continuous real world learning will be indispensable to optimizing patient outcomes and sustaining confidence in this rapidly evolving therapeutic class.
BLA / MAA: From Clinical Success to Global Approval
What Regulators Expect
- Integrated narrative linking clinical outcomes to CMC control
- Justification of specifications for:
- Drug-to-antibody ratio (DAR)
- Free payload limits
- Stability and shelf-life
- Clear labeling proposals addressing ADC-specific risks
- Risk management plans that reflect real-world use
Fortrea perspective: The strongest submissions demonstrate that clinical, CMC, and pharmacology teams have been aligned throughout development, not stitched together at the end.
Mapping ADC Regulatory Risk to Clinical Design Decisions
ADC Risk Area | Why Regulators Care | Clinical Design Response |
Free payload exposure | Drives systemic toxicity | Early and repeated PK sampling; conservative escalation |
Delayed toxicity | Often missed in short DLT windows | Extended safety monitoring and late follow‑up |
DAR variability | Impacts efficacy and safety | Consistent product supply and batch qualification |
ILD / pneumonitis | Increasing class concern | Baseline imaging, proactive monitoring, clear dose holds |
Dose justification | Project Optimus focus | Multiple dose levels in expansion cohorts |
Global Considerations: FDA vs EMA vs PMDA
FDA (United States):
Most explicit ADC expectations, including dedicated clinical pharmacology guidance. There is a strong emphasis on:
EMA (European Union)
- Applies monoclonal antibody and ICH frameworks to ADCs. The focus is on:
- Manufacturing robustness
- Long-term safety
- Early scientific advice strongly encouraged
PMDA (Japan)
- Conservative approach to:
- Starting dose
- Safety margins
- Often expects Japan-specific PK or bridging rationale
- High value placed on early consultation and alignment
Fortrea perspective: Global ADC programs benefit from region-specific strategy built into a single, coherent development plan, rather than reactive adjustments.
How Fortrea Supports ADC Sponsors
Fortrea collaborates with sponsors to:
- Integrate regulatory expectations into protocol design
- Optimize ADC dose-finding strategies aligned with global guidance
- Anticipate safety, pharmacology, and CMC questions before regulators ask them
- Execute global oncology trials with a focus on quality and efficient execution
Our experience across early- and late-phase ADC programs enables sponsors to move forward with greater clarity and preparedness when navigating the regulatory complexities.
Final Takeaway
ADCs demand more than strong science—they require strategic clinical development informed by regulatory insight. Sponsors who embed this early in the development are better positioned to navigate development efficiently, manage risk, and bring meaningful therapies to patients.
Learn more about antibody drug conjugate (ADC) clinical development and regulatory strategy.
Disclaimer:
This article is for general informational purposes only and does not constitute regulatory, scientific, or operational advice. Any timelines, performance metrics, or regulatory outcomes referenced are case specific and should not be interpreted as guarantees. Results may vary by study, jurisdiction, and study characteristics.
References
- Clinical Pharmacology Considerations for Antibody-Drug Conjugates: Guidance for Industry. FDA; March 2024. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-considerations-antibody-drug-conjugates-guidance-industry
- Clinical Pharmacology Considerations for Antibody-Drug Conjugates; Guidance for Industry; Availability. Federal Register Vol. 89, No. 42; March 1, 2024. Available at: https://www.federalregister.gov/documents/2024/03/01/2024-04375/clinical-pharmacology-considerations-for-antibody-drug-conjugates-guidance-for-industry-availability
- Guideline on Development, Production, Characterization and Specification for Monoclonal Antibodies and Related Products (EMA/CHMP/BWP/532517/2008). EMA; July 2016. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-production-characterisation-and-specification-monoclonal-antibodies-and-related-products-revision-1_en.pdf
- Bechtold-Peters K, et al. CMC Regulatory Considerations for Antibody-Drug Conjugates. Journal of Pharmaceutical Sciences. 2023;112(12):2965–2980. doi:10.1016/j.xphs.2023.09.007
- Chauhan V. Navigating Global Regulations for Antibody-Drug Conjugates (ADCs): A CMC Perspective. CASSS CMC Strategy Forum Japan; December 2023. Available at: https://www.casss.org
- Morgan C. CMC Regulatory Considerations for Antibody-Drug Conjugates. CASSS Summer Strategy Forum North America; July 2024. Available at: https://www.casss.org
- Cha KJ. Regulatory Considerations. In: Antibody-Drug Conjugates. AAPS Advances in the Pharmaceutical Sciences Series. Springer; 2025:153–175. doi:10.1007/978-3-031-83005-1_8